Кардиомиопатия дилатационная — описание, причины, симптомы (признаки), диагностика, лечение.
Краткое описание
Дилатационная кардиомиопатия (ДКМП) — первичное поражение сердца, характеризующееся расширением его полостей и нарушением сократительной функции. Статистические данные. Заболеваемость в мире составляет 3–10 случаев на 100 000 человек в год. Мужчины заболевают чаще женщин (2:1).
Код по международной классификации болезней МКБ-10:
- I42.0 Дилатационная кардиомиопатия
Причины
Этиология. Возникновение ДКМП связывают с взаимодействием нескольких факторов: генетических нарушений, экзогенных влияний, аутоиммунных механизмов.
• Экзогенные факторы •• Выявлена связь между перенесённым инфекционным миокардитом и развитием ДКМП ••• Установлено, что ДКМП может развиться после миокардита (в 15% случаев) в результате воздействия инфекционных агентов (энтеровирусы, боррелии, HCV, ВИЧ и др.). С помощью методики молекулярной гибридизации обнаружена энтеровирусная РНК в ядерной ДНК у больных с миокардитом и ДКМП ••• После инфекции, обусловленной вирусами Коксаки, может развиться сердечная недостаточность (даже через несколько лет) •• Получены убедительные данные о том, что токсическое воздействие алкоголя на миокард может привести к возникновению ДКМП •• В экспериментальных исследованиях воздействие этанола или его метаболита ацетальдегида вызывает уменьшение синтеза сократительных белков, повреждение митохондрий, образование свободных радикалов и повреждение кардиомиоцитов (наблюдают увеличение содержания тропонина Т в крови как признак поражения миокарда). Однако следует иметь ввиду, что тяжёлое поражение миокарда по типу ДКМП возникает лишь у части лиц (1/5), злоупотребляющих алкоголем •• • Хроническое воздействие этанола вызывает уменьшение синтеза белка, повреждение саркоплазматической сети и образование токсических эфиров жирных кислот и свободных радикалов. Кроме того, хроническое потребление алкоголя вызывает нарушения питания и всасывания, ведущие к дефициту тиамина, гипомагниемии, гипофосфатемии. Эти нарушения обусловливают изменение энергетического метаболизма клеток, механизма возбуждения — сокращения и усиливают дисфункцию миокарда.
• Аутоиммунные нарушения. Под воздействием экзогенных факторов белки сердца приобретают антигенные свойства, что стимулирует синтез АТ и провоцирует развитие ДКМП. При ДКМП обнаружено увеличение содержания в крови цитокинов, увеличенное количество активированных Т — лимфоцитов. Кроме того, обнаруживают АТ к ламинину, миозину тяжёлых цепей, тропомиозину, актину.
Генетические аспекты. Семейная ДКМП, в развитии которой генетический фактор, видимо, играет решающую роль, наблюдается в 20–30% всех случаев этой болезни. Выделены несколько видов семейных форм ДКМП с различными генетическими нарушениями, пенетрантностью и клиническими проявлениями.
• Кардиомиопатия семейная дилатационная: • тип 1B, CMD1B, CMPD1, FDC, 600884, 9q13; • тип 1C: CMD1C, CMPD3, 601493, 10q21 q23; • тип 2: CMPD2, 601494, 1q32; • с дефектом проведения, тип 2: CDCD2, CMPD2, 601154, 3p25 p22; • с дефектом проведения, тип 1: CMD1A, CDCD1, 115200 (a — актин сердечный), 1p11 q11.
• Кардиомиопатия X — сцепленная дилатационная (синдром Барта). Клинически: ДКМП, множественные миопатии, фиброэластоз эндокарда, сердечная недостаточность, нейтропения (остановка дифференцировки на стадии миелоцитов), задержка роста, пиодермии, дефектные митохондрии. Лабораторно: у ряда пациентов обнаруживают экскрецию с мочой 3 — метилглутарата.
Патогенез. Под действием экзогенных факторов уменьшается количество полноценно функционирующих кардиомиоцитов, что приводит к расширению камер сердца и нарушению сократительной функции миокарда. Полости сердца расширяются, что приводит к развитию систолической и диастолической дисфункции обоих желудочков). Развивается хроническая сердечная недостаточность.
• На начальных стадиях заболевания действует закон Франка–Старлинга (степень диастолического растяжения пропорциональна силе сокращения волокон миокарда). Сердечный выброс сохраняется также за счёт увеличения ЧСС и уменьшения периферического сопротивления при физической нагрузке.
• Постепенно компенсаторные механизмы нарушаются, увеличивается ригидность сердца, ухудшается систолическая функция и закон Франка–Старлинга перестаёт действовать •• Уменьшаются минутный и ударный объёмы сердца, увеличивается конечное диастолическое давление в левом желудочке и происходит дальнейшее расширение полостей сердца •• Возникает относительная недостаточность митрального и трёхстворчатого клапанов из — за дилатации желудочков и расширения клапанных колец •• Возникает компенсаторная гипертрофия миокарда в результате гипертрофии миоцитов и увеличения объёма соединительной ткани (масса сердца может превышать 600 г) •• Уменьшение сердечного выброса и увеличение внутрижелудочкового диастолического давления могут привести к уменьшению коронарной перфузии, следствием чего становится субэндокардиальная ишемия.
• Уменьшение сердечного выброса и снижение перфузии почек стимулируют симпатическую нервную и ренин — ангиотензиновую системы •• Катехоламины повреждают миокард, приводя к тахикардии, аритмиям и периферической вазоконстрикции •• Ренин — ангиотензиновая система вызывает периферическую вазоконстрикцию, вторичный гиперальдостеронизм, приводя к задержке ионов натрия, жидкости и развитию отёков, увеличению ОЦК и преднагрузки.
• Для ДКМП характерно формирование в полостях сердца пристеночных тромбов: в ушке левого предсердия, ушке правого предсердия, правом желудочке, левом желудочке.
Симптомы (признаки)
Клинические проявления. Проявления ДКМП включают застойную сердечную недостаточность, нарушения ритма и тромбоэмболии (возможно наличие как одного, так и всех трёх признаков). Заболевание развивается постепенно, но при отсутствии лечения (а часто даже и на фоне лечения) неуклонно прогрессирует. Жалобы длительное время могут отсутствовать.
• Жалобы •• Характерные для хронической сердечной недостаточности: одышка, слабость, утомляемость, сердцебиение, периферические отёки •• При расспросе больных нужно выяснить возможные этиологические моменты (семейный анамнез, вирусная инфекция, токсические воздействия, другие заболевания, в т.ч. и сердца).
• При декомпенсации отмечают признаки застоя в малом (одышка, хрипы в лёгких, ортопноэ, приступы сердечной астмы, «ритм галопа») и большом (периферические отёки, асцит, гепатомегалия) кругах кровообращения, сниженного сердечного выброса (снижение периферической перфузии в виде цианоза и холодной влажной кожи, низкое систолическое АД) и нейроэндокринной активации (тахикардия, периферическая вазоконстрикция).
• Одно из ранних проявлений ДКМП — пароксизмальная мерцательная аритмия ( как правило, быстро переходит в постоянную форму).
• При перкуссии сердца можно выявить расширение границ относительной сердечной тупости в обе стороны (кардиомегалия), при аускультации — систолические шумы относительной недостаточности трёхстворчатого и митрального клапанов. Характерно нарушение ритма в виде фибрилляции предсердий.
Диагностика
Инструментальные данные
• ЭКГ •• Признаки гипертрофии и перегрузки левого желудочка (депрессия сегмента ST и отрицательные зубцы T в I, aVL, V5. V6 ), левого предсердия •• У 20% больных ДКМП обнаруживают фибрилляцию предсердий •• Возможны нарушения проводимости, в частности блокада левой ножки пучка Хиса (до 80% больных), наличие которой коррелирует с высоким риском внезапной сердечной смерти (появление блокады левой ножки пучка Хиса связывают с развитием фиброзного процесса в миокарде) •• Характерно удлинение интервала Q–T и его дисперсия •• Реже возникает АВ — блокада.
• Мониторирование по Холтеру позволяет выявить угрожающие для жизни аритмии и оценить суточную динамику процессов реполяризации.
• ЭхоКГ позволяет выявить основной признак ДКМП — дилатацию полостей сердца с уменьшением фракции выброса левого желудочка. В допплеровском режиме можно обнаружить относительную недостаточность митрального и трёхстворчатого клапанов (может иметь место и относительная недостаточность аортального клапана), нарушения диастолической функции левого желудочка. Кроме того, при ЭхоКГ можно провести дифференциальную диагностику, выявить вероятную причину сердечной недостаточности (пороки сердца, постинфарктный кардиосклероз), оценить риск тромбоэмболии при наличии пристеночных тромбов.
• Рентгенологическое исследование помогает выявить увеличение размеров сердца, признаки лёгочной гипертензии, гидроперикарда.
• Радионуклидные методы исследования — диффузное снижение сократительной способности миокарда, накопление радионуклида в лёгких.
• МРТ позволяет выявить дилатацию всех отделов сердца, снижение сократительной способности миокарда левого желудочка, венозный застой в лёгких, структурные изменения миокарда.
Диагностика. Диагноз ДКМП ставят путём исключения других заболеваний сердца, проявляющихся синдромом хронической систолической сердечной недостаточности.
Дифференциальная диагностика. У ДКМП нет каких — либо патогномоничных клинических или морфологических маркёров, что затрудняет дифференциальную диагностику её со вторичными поражения миокарда известной природы (при ИБС, артериальной гипертензии, микседеме, некоторых системных заболеваниях и т.д.). Последние при наличии дилатации камер сердца называют вторичными кардиомиопатиями. Особенно трудной иногда бывает дифференциальная диагностика ДКМП с тяжёлым ишемическим поражением миокарда у относительно пожилых людей при отсутствии характерного болевого синдрома в виде стенокардии. При этом следует обращать внимание на наличие факторов риска атеросклероза, наличие атеросклеротического поражения аорты и других сосудов, но решающим могут быть данные коронарографии, позволяющей исключить стенозирующее поражение коронарных артерий. Тем не менее благодаря позитронной эмиссионной томографии миокарда появилась возможность очень точной дифференциальной диагностики между ДКМП и ишемической кардиомиопатией.
Лечение
Общая тактика. Лечение ДКМП заключается в адекватной коррекции проявлений сердечной недостаточности. В первую очередь необходимо ограничить количество потребляемой соли и жидкости. Также необходима коррекция возникающих нарушений ритма.
Лекарственная терапия
• Всем больным ДКМП при отсутствии противопоказаний необходимо назначать ингибиторы АПФ (каптоприл, эналаприл, рамиприл, периндоприл и др.). Препараты этой группы предупреждают прогрессирование сердечной недостаточности. При появлении задержки жидкости ингибиторы АПФ комбинируют с диуретиками, в основном фуросемидом.
• При тяжёлой сердечной недостаточности показано применение спиронолактона в дозе 25 мг/сут.
• Кроме того, может быть использован дигоксин, особенно при наличии мерцательной аритмии.
• Значительные трудности в лечении больных ДКМП возникают при наличии стойкой тахикардии и тяжёлых нарушений ритма сердца •• Терапия дигоксином в дозах более 0,25–0,375 мг/сут у таких больных быстро приводит к развитию гликозидной интоксикации даже при нормальной концентрации калия в сыворотке крови. В таких случаях целесообразно использование b — адреноблокаторов (бисопролола, карведилола, метопролола). Применение b — адреноблокаторов особенно показано при постоянной форме мерцательной аритмии. О благоприятном действии b — адреноблокаторов при ДКМП свидетельствуют результаты ряда клинических испытаний, которые подтвердили увеличение выживаемости больных под влиянием препаратов этой группы •• При сердечной недостаточности лучше всего изучена эффективность кардиоселективных препаратов метопролола и бисопролола, а также карведилола, блокирующего не только b -. но и a 1 — адренорецепторы. Блокада последних приводит к расширению сосудов.
• Антиагреганты — в связи с наклонностью к тромбообразованию целесообразно длительное применение антиагрегантов — ацетилсалициловой кислоты по 0,25–0,3 г/сут.
Хирургическое лечение — см. Недостаточность сердечная хроническая диастолическая, Недостаточность сердечная хроническая систолическая.
Осложнения. Наиболее частые осложнения ДКМП: артериальные и лёгочные тромбоэмболии (20% больных), нарушения ритма и проводимости сердца (30% больных), внезапная сердечная смерть, прогрессирующая сердечная недостаточность.
• Неблагоприятный прогноз имеют больные ДКМП при наличии следующих проявлений •• Симптомы сердечной недостаточности в покое (IV функциональный класс по Нью — Йоркской классификации) •• Выраженная дилатация левого или правого желудочка, выявленная при ЭхоКГ или рентгенологическом исследовании •• Сферическая форма левого желудочка по данным ЭхоКГ •• Низкая фракция выброса левого желудочка по данным ЭхоКГ •• Низкое систолическое АД •• Низкий сердечный индекс (менее 2,5 л/мин/м2) •• Высокое давление наполнения левого и правого желудочка •• Признаки выраженной нейроэндокринной активации — низкое содержание в крови ионов натрия, увеличенное содержание в крови норэпинефрина.
• 10 — летняя выживаемость больных с ДКМП в среднем составляет 15–30%. Смертность достигает 10% в год. При малосимптомном течении ДКМП 5 — летняя выживаемость больных не превышает 80%. У больных, госпитализированных по поводу хронической сердечной недостаточности, пятилетняя выживаемость составляет 50%. При рефрактерной сердечной недостаточности (IV функциональный класс по Нью — Йоркской классификации) выживаемость в течение 1 года не превышает 50%.
Особенности у детей. В первые 3 года жизни наиболее часто манифестируют наследственные и идиопатические формы ДКМП.
Беременность. При ДКМП, развившейся в период беременности или раннем послеродовом периоде, повторная беременность противопоказана.
Синонимы • Застойная кардиомиопатия • Конгестивная кардиомиопатия.
Сокращение. ДКМП — дилатационная кардиомиопатия.
МКБ-10 • I42.0 Дилатационная кардиомиопатия.
Дилатационная кардиомиопатия
Содержание:
Определение
Дилатационную кардиомиопатию характеризует расширение полостей сердца и прогрессирующая систолическая дисфункция миокарда, хотя практически при этом развивается и диастолическая дисфункция. Исходом данного заболевания часто бывает застойная сердечная недостаточность. Заболеваемость составляет 5-8 случаев на 100 тыс. населения. Представители негроидной расы и мужчины болеют в 3 раза чаще, чем европеоиды и женщины.
Причины
Хотя причина развития дилатационной кардиомиопатии по существу неизвестна, накопленные экспериментальные и клинические данные позволяют предполагать участие в ее патогенезе генетических, вирусных и аутоиммунных факторов. Тяжелые мутации генов, могут быть причиной наследственной дилатационной кардиомиопатии, тогда как определенные вирусы ответственны за развитие, спорадических случаев.
Возможные причины возникновения заболевания включены в следующий перечень:
- наследственная (может быть непосредственной причиной более чем в 25% всех случаев);
- миокардиты (инфекционные, аутоиммунные, токсические);
- метаболическая (при гемохроматозе, тиреотоксикозе);
- связанная с влиянием диетических факторов (дефицит тиамина, болезнь бери-бери);
- на фоне персистирующей тахикардии (тахимиопатия).
Диагноз дилатационной кардиомиопатии по существу диагноз исключения. Потенциально обратимые причины заболевания, такие как поражение коронарных артерий, патология клапанов, врожденные пороки, должны быть своевременно выявлены и устранены. Особое внимание необходимо уделить оценке диеты и потребления алкоголя, как с изменением данных факторов в некоторой степени возможен регресс заболевания.
Симптомы
Клинически заболевание может сразу проявиться острым отеком легких, ТЭЛА или артерий большого круга кровообращения. Возможна внезапная смерть. Но чаще у пациентов выявляют симптомы застойной сердечной недостаточности — одышку, усиливающуюся при физической нагрузке, ортопноэ, пароксизмальную ночную одышку, общую слабость. Достаточно часто характерно развитие аритмий (ФП, особенно при употреблении большого количества алкоголя), которые могут привести к ЖТ и внезапной остановке сердца.
Диагностика
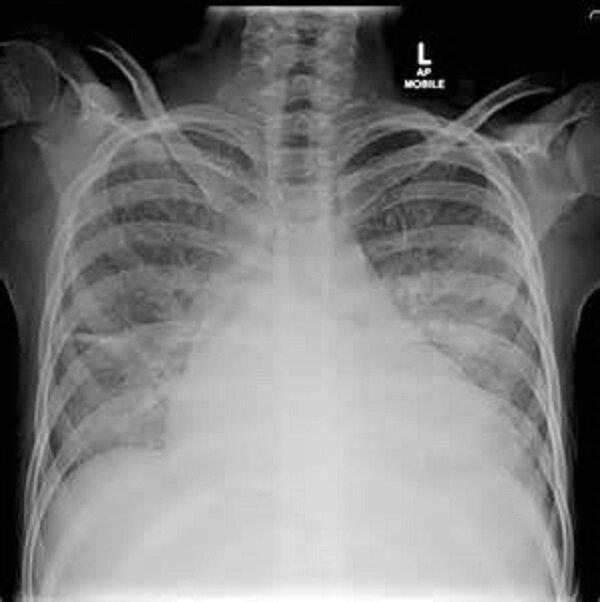 Диагноз устанавливают на основании осмотра, ЭКГ, данных рентгенографии грудной клетки и ЭхоКГ.
Диагноз устанавливают на основании осмотра, ЭКГ, данных рентгенографии грудной клетки и ЭхоКГ.
ЭКГ позволяет выявить гипертрофию левого желудочка, предшествующий ИМ, аритмии. Часто встречают синусовую тахикардию с характерными неспецифическими изменениями зубца Т и отсутствием увеличения зубца R в передних грудных отведениях.
Рентгенография органов грудной клетки может выявить увеличение размеров сердца и отек легких (венозное полнокровие верхних долей, интерстициальный отек, плевральный выпот и линии Керли).
ЭхоКГ позволяет точно оценить размеры камер, функции сердца и особенно клапанов. Характерна дилатация обоих желудочков с нарушением подвижности межжелудочковой перегородки (БЛНПГ). Возможно обнаружение пристеночного тромба в одном или обоих желудочках. Можно обнаружить в полости перикарда скопление небольшого количества жидкости. ФВ и фракция систолического укорочения левого желудочка снижены, дилатация желудочков является причиной развития митральной и трикуспидальной недостаточности.
Нагрузочные пробы с измерением (либо без) максимального потребления вдыхаемого кислорода позволяют оценить функциональный резерв. Холтеровское мониторирование способствует выявлению жизнеугрожающих желудочковых аритмий.
Катетеризацию сердца следует выполнять с осторожностью у пациентов с ухудшением функции левого желудочка, так как данная процедура может спровоцировать развитие острого отека легкого или тромбоэмболию пристеночным тромбом. Катетеризация сердца показана для:
- исключения значимой коронарной патологии;
- оценки тяжести митральной недостаточности и измерения давления в легочной артерии;
- биопсии желудочков в настоящее время используется только по строгим показаниям и позволяет доказать наличие острого миокардита (выявление лимфоцитарной инфильтрации); проведение ПЦР для определения генома вируса оказалось малоэффективным.
Профилактика
Лечение в основном симптоматическое, направленное на улучшение прогноза, и соответствует таковому при хронической сердечной недостаточности. Медикаментозное лечение направлено на коррекцию нарушений и восстановление гомеостаза нейрогуморальных систем — симпатической и ренин-ангиотензин-альдостероновой.
Диуретики. Применение диуретиков, в частности петлевых, вызывает уменьшение признаков периферического и легочного застоя. При таком лечении важно контролировать концентрацию электролитов плазмы, так как нарушение их баланса приводит к развитию гипокалиемии и повышению уровня мочевины. Коррекция концентрации калия может проводиться путем дополнительного назначения калийсберегающих диуретиков. Спиронолактон — прямой антагонист альдостерона и способен блокировать его действие в отношении задержки воды и соли.
Вазодилататоры. Действие ингибиторов АПФ изучено во многих крупных исследованиях. Показано, что препараты данной группы не только уменьшают выраженность симптомов заболевания, но и улучшают прогноз у пациентов с заболеваниями сердца, включая даже больных на бессимптомной стадии. Ранее в связи с гипотензией в ответ на введение первой дозы было предпочтительнее начинать терапию в стационарных условиях, но в настоящее время так поступают лишь с больными с уменьшением объема внутрисосудистой жидкости вследствие применения диуретиков в больших дозах. Побочные эффекты ингибиторов АПФ включают сухой кашель. вероятнее всего, из-за увеличения уровня брадикинина, ангионевротический отек (редко). Препараты данного класса следует с осторожностью назначать больным с рено-васкулярной патологией.
Антагонисты рецептора ангиотензина можно использовать как альтернативу у больных, плохо переносящих ингибиторы АПФ и имеющих реноваскулярную патологию.
β-Адреноблокаторы. Хотя β-адреноблокаторы оказывают отрицательный инотропный эффект, их использование улучшает симптомы и прогноз ввиду повышения диастолического наполнения желудочков и уменьшения частоты развития аритмий. Лечение должно начинаться малыми дозами с последующим титрованием. β-Адреноблокаторы не следует назначать пациентам с выраженными проявлениями хронической сердечной недостаточности, их необходимо отменять при декомпенсации состояния пациента.
Антиаритмики. К сожалению, применение антиаритмических препаратов не привело к уменьшению случаев внезапной коронарной смерти у пациентов с дилатационной кардиомиопатией. ФП при дилатационной кардиомиопатии встречают часто, при ее развитии необходимо контролировать частоту желудочковых сокращений с помощью соответствующих препаратов. Тем не менее, длительное поддержание синусового ритма с помощью антиаритмической терапии маловероятно, поэтому ее продолжительность необходимо ограничить.
Антикоагулянты. Пациенты с дилатационной кардиомиопатией предрасположены и возникновению тромбоэмболических осложнений. Поэтому независимо от наличия аритмии необходимо назначение соответствующей терапии варфарином.
 Нефармакологические подходы в лечении. Обратимые причины дилатационной кардиомиопатии (ИБС, пороки клапанов) подлежат коррекции. Реваскуляризация миокарда при тяжелой ИБС и сопутствующей дилатационной кардиомиопатии может значительно улучшить прогноз заболевания. При отсутствии эффекта от массивной медикаментозной терапии у тяжелых пациентов необходимо рассмотреть целесообразности выполнения ортотопической пересадки сердца. Однако ограниченное число донорских органов до сих пор является препятствием для широкого применения данной методики. В связи с этим большое количество пациентов умирают, так и не дождавшись своей очереди. Поэтому большой интерес представляет использование органов от представителей других видов – ксенотрансплантатов. На пути этой методики стоят многочисленные технические трудности. Применение механических устройств для поддержания жизненных функций организма (искусственного сердца) находится в стадии изучения и разработки.
Нефармакологические подходы в лечении. Обратимые причины дилатационной кардиомиопатии (ИБС, пороки клапанов) подлежат коррекции. Реваскуляризация миокарда при тяжелой ИБС и сопутствующей дилатационной кардиомиопатии может значительно улучшить прогноз заболевания. При отсутствии эффекта от массивной медикаментозной терапии у тяжелых пациентов необходимо рассмотреть целесообразности выполнения ортотопической пересадки сердца. Однако ограниченное число донорских органов до сих пор является препятствием для широкого применения данной методики. В связи с этим большое количество пациентов умирают, так и не дождавшись своей очереди. Поэтому большой интерес представляет использование органов от представителей других видов – ксенотрансплантатов. На пути этой методики стоят многочисленные технические трудности. Применение механических устройств для поддержания жизненных функций организма (искусственного сердца) находится в стадии изучения и разработки.
Сердечная ресинхронизирующая терапия (СРТ) направлена ни улучшение гемодинамических показателей сердца за счет одновременной стимуляции желудочков искусственным водителем ритма. В крупных исследованиях было доказано увеличение функциональных способностей пациентов и даже небольшое снижение смертности.
Онлайн консультация врача
Идиопатическая дилатационная кардиомиопатия
Что такое Идиопатическая дилатационная кардиомиопатия —
Идиопатическая дилатационная кардиомиопатия (ИДКМП) — диффузное заболевание миокарда неизвестной этиологии, характеризующееся дилатацией всех камер сердца с нарушением его систолической функции. Заболеваемость идиопатической дилатационной кардиомио-патией составляет 5-8 случаев на 100000 населения в год, причем наблюдается тенденция к увеличению этого показателя. Распространенность заболевания достигает 13-36 на 100000 населения. Вероятно, истинная заболеваемость выше вследствие неучтенности случаев, протекающих определенное время без клинической симптоматики или с незначительными клиническими проявлениями. Примерно 25% всех случаев ХСН обусловлены идиопатической дилатационной кардиомиопатией.
Что провоцирует Идиопатическая дилатационная кардиомиопатия:
Причина болезни не установлена. Goodwin (1973) сформулировал положение о полиэтиологичности заболевания. В настоящее время наиболее широко обсуждаются несколько гипотез развития ИДКМП. О большой роли генетических факторов свидетельствует тот факт, что семейный характер заболевания прослеживается у 20-25% пациентов, причем течение семейной формы заболевания является наиболее злокачественным. Наличие семейного характера заболевания свидетельствует о вкладе генетических факторов в его развитие, что подтверждается морфологическими, клиническими признаками, кардиогемодинамическими нарушениями при семейной и изолированной ИДКМП.
Известны четыре типа наследования ИДКМП: аутосомно-доминантный, аутосомно-рецессивный, сцепленный с Х-хромосомой и через митохондриальную ДНК. Наиболее распространенным является аутосомно-доминантный с передачей мутантных генов. Дилатационная кардиомиопатия с аутосомно-доминантным наследованием развивается в возрасте 20-30 лет и характеризуется прогрессирующей СН и тяжелыми аритмиями. В семьях этих больных идентифицированы пять локусов с локализацией мутации в девятой (9ql3-q22), первой (Iq32) и десятой хромосоме (10q21-10q23). При наличии у больного последней мутации наряду с дилатационной кардиомиопатией развивается пролапс митрального клапана. У больных старше 30 лет чаще обнаруживаются изменения первой хромосомы, а течение ИДКМП характеризуется изменением атриовентрикулярной и внутрижелудочковой проводимости, выраженными иммунологическими нарушениями. У многих больных с семейной ИДКМП выявлены мутации в шестой хромосоме (6q23 в области 3-сМ), что сопровождается нарушениями проводимости и дефектами мышечной ткани. У некотрых больных ИДКМП обнаружены мутации гена ламина с нарушением атриовентрикулярной проводимости. Дилатационная кардиомиопатия с аутосомно-рецессивным типом наследования встречается значительно реже. Генетический локус, ответственный за развитие заболевания, пока не установлен.
Вариант семейной дилатационной кардиомиопатии, сцепленный с полом (с Х-хромосомой) встречается в двух формах. Первая форма (синдром Барта) развивается в детском возрасте. При этой форме установлены 4 мутации в гене G4,5, который находится на длинном плече Х-хромосомы (Xq28) и кодирует синтез и функцию белков «тафазинов», входящих в состав структурных белков мембран. Синдром Барта характеризуется сочетанием дилатационной кардиомиопатии с отставанием детей в росте, миопатией, нейтропенией, аминоацидурией. Больные умирают рано, часто от сепсиса.
Вторая форма дилатационной кардиомиопатии, сцепленной с Х-хромосомой, развивается в более старшем возрасте, характеризуется быстрым прогрессирующим течением, миопатией, повышением содержания в крови креатинфосфокиназы. Генетический дефект при этом заболевании локализуется в Х-хромосоме — Хр21 и заключается в делеции гена, расположенного в премоторном регионе в первом экзоне и контролирующего синтез белка дистрофина — компонента цитоскелета миоцитов. В миокарде значительно снижается количество дистрофина и а-дистрогликана, гликопротеина ассоциированного с дистрофином.
Дистрофин входит в состав дистрофин-ассоциированного гликопротеинового комплекса и обеспечивает связь актинового цитоскелета миоцитов с внеклеточным матриксом. Актиновый цитоскелет связан также с сократительным аппаратом миоцита через миокардиальный LIM (Lin-11, Isl-1, Мес-3) протеин (MLP). Этот протеин играет важную роль в дифференциации и пролиферации клеток. Мутация гена дистрофина, других белков дистрофин-ассоциированного гликопротеинового комплекса, протеина MLP приводит к повреждению и гибели кардиомиоцитов, развитию кардиомиопатии.
В последние годы обнаружены митохондриальные кардиомиопатии при семейной, спорадической формах заболевания. Вместе с тем роль мутаций митохондриальной ДНК окончательно не выяснена. При ультраструктурном и иммуноцитохимическом изучении биоптатов миокарда обнаруживается патология митохондрий в виде концентрических и тубулярных кист, снижающих антиферментную активность митохондрий.
Митохондриальные синдромы включают дилатационную кардиомиопатию как составную часть клинической картины. К этим синдромам относятся: MELAS-синдром (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes — митохондриальная энцефалопатия, миопатия, лактацидоз и инсультоподобные эпизоды); MERRF-синдром (Myoclonic Epilepsy with Ragged Red Fibres — миоклональная эпилепсия с рваными мышечными волокнами) включает митохондриальную миопатию, миоклонии, большие эпилептические припадки, деменцию, атаксию, тугоухость; синдром Кирнса-Сейра обусловлен делецией мутантного гена митохондрий, характеризуется прогрессирующей наружной офтальмоплегией, пигментной дегенерацией сетчатки, атриовентрикулярной блокадой 1-3 степени. У больных ИДКМП часто обнаруживается DD-генотип ангиотензинпревращающего фермента, что рассматривается как маркер предрасположенности к развитию болезни.
Изучается вирусная этиология ИДКМП. Предполагается, что перенесенный вирусный миокардит инициирует аутоиммунный воспалительный процесс, который трансформируется в делатационнуб кардиомиопатию. В пользу этой гиротезы свидетельствуют наличие симптоматики вирусоподобного заболевания с лихорадкой, предшествовавшего развитию ИДКМП (20-25%); смена воспаления миокарда признаками дилатационной кардиомиопатии в прижизненных биоптатах больных, перенесших вирусный миокардит (12-52%); выявление у больных ИДКМП в диагностически значимых титрах антител к кардиотропным вирусам (энтеровирус Коксаки группы В); обнаружение в 12-67% биоптатов миокарда при ИДКМП энтеровирусной РНК, комплементарной к РНК вирусов Коксаки; отсутствие в биоптатах миокарда признаков воспаления; трансформация экспериментального энтеровиусного миокардита через 6-12 месяцев в морфологические признаки ИДКМП.
В качестве этиологического фактора ИДКМП могут выступать метаболические нарушения миокарда — врожденные или приобретенные в течение жизни метаболические дефекты, которые приводят к дилатации сердца и его недостаточности. При дефиците карнитина поступление жирных кислот в митохондрии и их окисление значительно снижаются. Это сопровождается их накоплением в цитоплазме, дефицитом энергии, расширением полостей сердца и развитием ХСН. При ИДКМП в миокарде обнаруживаются патологические изменения обмена веществ: повышенная продукция оксида азота стимулирует апоптоз кардиомиоцитов; снижение активности Са-АТФазы саркоплазматического ретикулума; дефицит цитоскелетного белка кардиомиоцитов метавинкулина. Указанные изменения метаболизма в миокарде некоторыми исследователями рассматриваются как этиологический фактор рассматриваемого заболевания. У больных ИДКМП обнаружены значительные нарушения клеточного и гуморального иммунитета, которые способствуют прогрессированию заболевания и недостаточности кровообращения.
Обнаруживаются циркулирующие в крови антитела к тяжелым цепям миозина, р-адренорецепторам, мускариновым рецепторам, ламинину, митохондриальным белкам. Особая роль в патогенезе ИДКМП придается антителам к Р-адренорецепторам миокарда (30-40%). Это обусловлено тем, что циркулирующие аутоантитела к р-адренорецепторам уменьшают плотность р-адренорецепторов в миокарде, снижают их функциональную активность и количество, уменьшают таким образом кардиоинотропное влияние симпатической нервной системы и способствуют прогрессированию кардиальной дисфункции, дезадаптивному ремоделированию ЛЖ.
Большое патогенетическое значение имеют специфические антитела к ферменту внутренней мембраны митохондрий сердца, осуществляющему перенос АТФ и АДФ между цитоплазмой и матриксом митохондрий (57%). Антитела к адениннуклеотидному транслокатору обладают перекрестной реактивностью к белкам кальциевых каналов и, вступая во взаимодействие с ними, приводят к чрезмерному поступлению ионов Са++ в кардиомиоциты, вызывают перегрузку кардиомиоцитов кальцием, их повреждение, лизис. Снижение транспорта АТФ из митохондрий к сократительным белкам кардиомиоцита приводит к снижению коронарного кровотока, сердечного выброса, потребления миокардом кислорода.
Значительные изменения клеточного иммунитета характризуются снижением активности натуральных киллеров, нарушением функции Т-лимфоцитов-хелперов, повышением активности Т-лимфоцитов-хелперов, увеличением продукции интерферона, фактора некроза опухоли-альфа, интерлейкина-2. В результате указанных сдвигов развиваются аутоиммунные реакции к белкам миокарда. Продуцируемые аутоантитела к миокарду вместе с провоспалительными цитокинами вызывают повреждение миокарда, способствуют развитию и прогрессированию ИДКМП. Как один из механизмов патогенеза ИДКМП следует рассматривать дисфункцию симпатической нервной системы.
Одним механизмов патогенеза ИДКМП является апоптоз (запрограммированная смерть клеток). Морфологическими признаками апоптоза являются: сморщивание клетки, конденсация и фрагментация ядра, разрушение цитоскелета, буллезное выпячивание клеточной мембраны. Характерной особенностью апоптоза является уничтожение клеток без развития воспаления. Целостность мембраны умирающей клетки сохраняется до полного завершения апоптоза. После окончания апоптоза расположенные вблизи фагоциты поглощают оставшиеся фрагменты клетки. Апоптоз регулируется семейством генов BCL-2, расположенных на 18 хромосоме. Гены BCL-2, C-FES тормозят, а гены ВАХ, ВАК, BID, Р-53, C-MYC, APO-1/Fas — стимулируют апоптоз.
В регуляции апоптоза принимают участие ростовые факторы, различные цитокины, половые гормоны. Апоптоз кардиомиоцитов активируется преимущественно фактором некроза опухоли, интерлейкинами-1, 4, g-интерфероном, продуктами перекисного окисления липидов, гипоксией. Распад клетки, предназначенной для апоптоза, происходит под влиянием ферментов цистеиновых протеаз. Запускают апоптоз проапоптотические сигналы двух типов: повреждение ДНК клетки какими-либо факторами и активация рецепторов «региона клеточной смерти» (Fas-R, TNF-R). Эти рецепторы являются мембраносвязанными белками, относящимися к семейству туморнекротических рецепторов. Повреждение ДНК вызывает активацию проапоптотических генов. Активация этих генов повышает проницаемость митохондрий клетки и выход в цитоплазму цитохрома С, АТФ, апоптозиндуцирующего фактора и ДНКазы.
Симптомы Идиопатической дилатационной кардиомиопатии:
Идиопатической дилатационной кардиомиопатией болеют чаще мужчины в возрасте 30-45 лет (85%). Заболевание встречается в 3 раза чаще среди людей черной расы. Около 30% больных указывают, что развитию клинических проявлений предшествовала острая респираторная вирусная инфекция, ангина, внебольничная пневмония и другие инфекционные заболевания.
Особенности течения
Начало заболевания постепенное, мало заметное (70-80%). Больные могут отмечать небольшую слабость и одышку, но не придают этим симптомам большого значения, расценивая их как следствие напряженной, длительной работы, отсутствия достаточного отдыха. Однако с течением времени, спустя несколько месяцев или 2-10 лет развивается выраженная симптоматика СН, обнаруживается кардиомегалия. Вместе с тем у 20% больных отмечается подострое начало ИДКМП с быстрым развитием признаков СН. Описан редкий вариант начала ИДКМП, характеризующийся развитием недостаточности кровообращения во время острой вирусной инфекции. При таком начале заболевания вирусная инфекция является провоцирующим фактором, выявляющим скрыто протекавшую дилатационную кардиомиопатию. Иногда ИДКМП дебютирует тромбоэмболией (в легкие, головной мозг, периферические сосуды) или аритмиями.
Существует семейно-генетическая форма заболевания, и анализ анамнеза позволяет ее своевременно распознать. Для семейной формы ИДКМП характерно быстрое начало и неуклонно прогрессирующее течение заболевания, наличие двух или более случаев дилатационной кардиомиопатии в одной семье; случаи документированной внезапной смерти в возрасте до 35 лет среди родственников первой степени родства. Клиническая картина ИДКМП характеризуется СН, нарушениями сердечного ритма, кардиомегалией, тромбоэмболическим синдромом.
На общую слабость, снижение работоспособности, одышку, ощущение перебоев, боли в сердце. Одышка и слабость усиливаются по мере прогрессирования заболевания. Одышка обусловлена левожелудочковой недостаточностью. Вначале одышка появляется при физической нагрузке, затем беспокоит больных в покое. Левожелудочковая недостаточность обусловлена нарушением сократительной мпособности миокарда ЛЖ. При присоединении правожелудочковой недостаточности больные предъявляют жалобы на появление отеков голеней и стоп, боли в области правого подреберья.
Кардиалгии неинтенсивные, носят кратковременный или достаточно длительный характер (25-50%) и обусловлены растяжением перикарда вследствие дилатации сердца, несоответствием между возросшей потребностью дилатированного ЛЖ в кислороде и ограниченными возможностями коронарного кровотока, поражением системы микроциркуляции миокарда и развитием его ишемии преимущественно в субэндокардиальных отделах, сопутствующей ИБС у лиц пожилого возраста (25%). У многих больных возможно развитие интенсивных болей в грудной клетке вследствие ТЭЛА или в левом подреберье при тромбоэмболии в селезеночную артерию. Тромбоэмболия почечной артерии вызывает развитие интенсивных болей в поясничной области.
Объективное исследование
При осмотре обращают на себя внимание одышка, акроцианоз, вынужденное сидячее или полусидячее положение, пастозность или выраженные отеки в области нижних конечностей, набухание шейных вен. При выраженной трикуспидальной недостаточности определяется пульсация яремной вены.
Исследование сердечно-сосудистой системы
Пульс часто аритмичный и сниженной амплитуды, при тяжелой степени сердечной недостаточности — нитевидный. При перкуссии сердца отмечается расширение всех границ относительной тупости сердца, более выражено расширение левой границы. Характерными аускультативными признаками являются: ослабление тонов сердца (преимущественно I тона), выслушивание III и IV тонов, протодиастолический, реже — пресистолический ритм галопа, систолический шум над верхушкой сердца и мечевидным отростком в результате дилатации ЛЖ и ПЖ, формирования относительной митральной, трикуспидальной недостаточности. При развитии застойных явлений в малом круге кровообращения выслушивается акцент II тона над легочной артерией. Наиболее частыми нарушениями сердечного ритма (60-65%) являются экстрасистолия (90%), мерцательная аритмия, пароксизмальная тахикардия (30%). Артериальное давление обычно снижено или нормальное.
Исследование легких
Обнаруживаются укорочение перкуторного звука и ослабление везикулярного дыхания в нижних отделах легких, здесь же прослушиваются крепитация, мелкопузырчатые хрипы. Развитие недостаточности кровообращения сопровождается увеличением и болезненностью печени, тотальная СН сопровождается развитием асцита.
Тромбоэмболические осложнения
Важнейшей клинической особенностью ИДКМП является тромбоэмболический синдром (40-77%). Одинаково часто (30-50%) встречаются тромбы в левых и правых отделах сердца, которые являются источником эмболии в большом и малом кругах кровообращения. Чаще всего развивается ТЭЛА (50%), тромбоэмболия почечных артерий (20%), селезеночной артерии (11%), артерий нижних конечностей и головного мозга (5-6%). Наиболее часто тромбоэмболии развиваются в первые три года болезни, к 10 году болезни наблюдается у 30-40% больных.
Тромбоэмболия почечной артерии проявляется интенсивными болями в поясничной области, микрогематурией, подъемом уровня АД, повышением температуры тела, резкой болезненностью при пальпации живота в проекции почки, картиной инфаркта почки при ультразвуковом исследовании. Тромбоэмболия селезеночной артерии характеризуется сильной болью в области левого подреберья, повышением температуры тела, иногда появлением шума трения брюшины при аускультации над поверхностью селезенки, картиной инфаркта селезенки при ультразвуковом исследовании.
Тромбоэмболия артерий ног проявляется внезапной острой болью, распространяющейся на весь отдел конечности дистальнее уровня закупорки артерии. Далее появляются онемение, бледность, похолодание конечности, снижается мышечная сила, больной теряет способность двигать ногой. При объективном обследовании отмечаются исчезновение пульса на магистральных артериях, побледнение, цианоз, снижение чувствительности кожи. Тромбоэмболия артерий головного мозга характеризуется внезапной потерей сознания, асимметрией носогубных складок, развитием парезов, других очаговых неврологических симптомов в зависимости от локализации тромбоэмболии.
В последнее десятилетие тромбоэмболии артерий прижизненно диагностируются в 15-45% случаев, потому что довольно часто протекают без клинической симптоматики или под масками пневмонии при ТЭЛА, усугубления симптомов СН, мочевого синдрома.
Течение идиопатической дилатационной кардиомиопатии весьма вариабельно и отличается индивидуальными особенностями у различных больных. Все же в большинстве случаев заболевание протекает тяжело, и средняя продолжительность жизни больных с момента появления первых клинических признаков колеблется от 3.4 до 7.1 лет (Ikram и соавт. 1987). Dec и Fuster (1994) указывают, что У4 всех впервые выявленных больных с идиопатической дилатационной кардиомиопатией умирает в течение года, а ‘/2 больных — в течение ближайших 5 лет.
Характеризуют прогноз при идиопатической дилатационной кардиомиопатии следующим образом: большая часть летальных исходов наблюдается в течение первых двух лет, пик летальности приходится на срок 6 месяцев — 1 год от появления первых симптомов недостаточности кровообращения. Летальность за первый год составляет 20 — 35%, за 3 года — 35 — 50%, за 5 лет — 50 — 70%, через 10 лет остаются в живых 25 — 30% больных. Основными причинами смерти больных являются: внезапная смерть от фибрилляции желудочков (‘/3
‘/2 умерших); застойная недостаточность кровообращения (летальных исходов); массивная тромбоэмболия легочной артерии (12 — 18% летальных исходов).
Большинство кардиологов выделяют 3 варианта течения идиопатической дилатационной кардиомиопатии: неуклонное ухудшение состояния больного, завершающееся смертью в течение 1 — 2 лет; прогрессирование с последующей стабилизацией и даже улучшением с последующим ухудшением состояния; длительность течения больше 15 лет без выздоровления; медленно прогрессирующее течение с внезапной смертью. По данным Semigran и соавт. (1994), у ‘/4 больных с недавно выявленной идиопатической дилатационной кардиомиопатией возможно спонтанное улучшение состояния.
Тем не менее, в подавляющем большинстве случаев прогноз при идиопатической дилатационной кардиомиопатии неблагоприятен в связи с прогрессирующей сердечной недостаточностью, рефрактерной к лечению, и тяжелыми, нередко фатальными нарушениями сердечного ритма и тромбоэмболиями. По данным Е. Н. Амосовой (1990), основными предикторами неблагоприятного прогноза являются застойная сердечная недостаточность III и IV классов (по Нью-Йоркской классификации), тромбоэмболии в анамнезе, увеличение конечного диастолического давления в левом желудочке более 20 мм рт. ст. и конечного диастолического объема более 150 см3/м2. Frahwald и соавт. (1994) к прогностически неблагоприятным факторам относят также наличие протодиасто-лического ритма галопа, пожилой возраст, резко выраженную ди-латацию сердца.
Ограничение физической активности больных и сниженное максимальное поглощение кислорода (ниже 10 — 12 мл/кг/мин) являются реальными предикторами летального исхода и учитываются при решении вопроса о необходимости трансплантации сердца. Pellicia и соавт. (1994) предлагают учитывать при оценке прогноза идиопатической дилатационной кардиомиопатии такой специфический признак, обнаруживаемый в эндомиокардиальных биоптатах, как отсутствие внутриклеточных миофиламентов (показатель неблагоприятного прогноза).
Плохое прогностическое значение имеют также частая желудочковая экстрасистолия и эпизоды пароксизмальной желудочковой тахикардии. Важнейшими прогностическими факторами считают выраженные клинические проявления, низкие величины фракции выброса левого желудочка и сердечного индекса, сложные желудочковые эктопические нарушения ритма, гипонатриемию, высокое содержание в крови норадреналина и предсердного натрийуретического гормона.
Диагностика Идиопатической дилатационной кардиомиопатии:
ЛАБОРАТОРНО-ИНСТРУМЕНТАЛЬНАЯ ДИАГНОСТИКА
Приведенная выше клиническая симптоматика идиопатической дилатационной кардиомиопатии является неспецифической и может наблюдаться не только при этом заболевании, но и при миокардитах и других видах кардиомиопатии, что затрудняет своевременную диагностику идиопатической дилатационной кардиомиопатии. Указанное обстоятельство делает важным и целесообразным использование лабораторно-инструментальных исследований в своевременной диагностике дилатационной кардиомиопатии.
Общий анализ крови
У большинства больных без существенных изменений.
Биохимический анализ крови
Не выявляет отклонений от нормы. У некоторых больных может обнаруживаться повышение содержания в крови креатинфосфокиназы и ее изоэнзима MB, что может быть обусловлено продолжающимися выраженными дистрофическими изменениями в миокарде, прогрессирующим повреждением миокарда, развитием в нем явлений некроза кардиомиоцитов. Повышение активности в сыворотке крови креатинфосфокиназы имеет неблагоприятное прогностическое значение, так как всегда ассоциируется с тяжелой и прогрессирующей застойной сердечной недостаточностью. М. Ю. Самсонов и соавт. (1991) обнаружили повышение содержания в крови проколлагена III типа при дилата-ционной кардиомиопатии, что отражает выраженность развития фиброза в миокарде.
Коагулограмма
У многих больных выявляет повышение свертывающей активности крови и признаки диссеминированного внутрисосудистого свертывания (в частности, высокий уровень в крови плазменного D-димера).
Иммунологические исследования
У некоторых больных обнаруживаются снижение количества и функциональной активности Т-лимфоцитов-супрессоров и повышение количества Т-лимфоцитов-хелперов, увеличение концентрации отдельных классов иммуноглобулинов, однако, эти изменения очень вариабельны и не имеют большого диагностического значения.
Электрокардиография
Характерной особенностью ЭКГ при дилатационной кардиомиопатии является нарушение сердечного ритма. А. Ю. Ибрагимов (1989) и Ю. И. Новиков (1988) указывают, что синусовый ритм при дилатационной кардиомиопатии регистрируется у 60 — 65% больных, а различные аритмии — у 40 — 35% больных. Однако эти авторы проводили однократные ЭКГ-исследования.
По данным Е.Н.Амосовой (1999) и П. X. Джанашия и соавт. (2000), холтеровское ЭКГ-мониторирование выявляет разнообразные нарушения практически у 100% больных дилатационной кар-диомиопатией, при этом наиболее часто регистрируются желудочковые экстрасистолы (почти у всех больных), короткие «пробежки» желудочковой тахикардии отмечаются у 15 — 60%, пароксизмы желудочковой тахикардии — у 5 — 10% больных. Существует мнение, что на частоту желудочковых аритмий степень тяжести сердечной недостаточности и давность заболевания значительного влияния не оказывают.
Приблизительно у 25-35% больных идиопатической дилатационной кардиомиопатией наблюдается мерцательная аритмия. Около 30 — 40% больных имеют атриовентрикулярные блокады различной степени, а у 40 — 50% наблюдается полная блокада левой ножки пучка Гиса или ее передней ветви. В то же время блокада правой ножки пучка Гиса считается нехарактерной для идиопатической дилатационной кардиомиопатии и встречается редко.
Полная блокада левой ножки пучка Гиса обычно ассоциируется со значительно более выраженными сердечной недостаточностью и дилатацией левого желудочка. Характерны также неспецифические изменения фазы реполяри-зации в виде снижения амплитуды зубца Т или даже его негативности (обычно отрицательный зубец Т несимметричный) в нескольких грудных отведениях, причем эти изменения стабильны или малодинамичны и часто сопровождаются депрессией интервала ST.
Приблизительно у 70% больных на ЭКГ имеются признаки гипертрофии миокарда левого желудочка и левого предсердия. В 1994 г. Momijama и соавт. на основании детального изучения ЭКГ сумели выявить ЭКГ-признаки, наиболее характерные для дилатационной кардиомиопатии и, таким образом, отличающие ее от кардиомиопатии и кардиомегалий другой природы: наибольшая амплитуда зубца R в отведении V6 и наименьшая — в отведениях I, II или III; отношение высоты зубца R в отведении V6 к амплитуде наибольшего зубца R в отведениях I, II или III > 3 (у 67% больных с дилатационной кардиомиопатией). Второму признаку, по мнению Momijama и. соавт. (1994), следует уделять большое внимание, так как он встречается лишь у 4% больных пороками сердца и у 8% больных с артериальной гипер-тензией, и никогда не наблюдается при ишемической кардиомио-патии и у здоровых лиц. У больных дилатационной кардиомиопа-тией отношение RV6 к наибольшему R в отведениях I, II или III прямо пропорционально дилатации левого желудочка.
У некоторых больных идиопатической дилатационной кардио-миопатией могут обнаруживаться патологические зубцы Q, что может создавать трудности в дифференциальной диагностике с ишемической кардиомиопатией. Чаще зубцы Q выявляются в отведениях I, V5, V6 и обусловлены очаговым или диффузным кардиосклерозом при дилатационной кардиомиопатии.
Эхокардиография
В настоящее время эхокардиографическое исследование является важнейшим неинвазивным методом диагностики идиопатической дилатационной кардиомиопатии. К эхокардиографическим признакам дилатационной кардиомиопатии относятся: дилатация всех полостей сердца (преимущественно желудочков, больше левого); практически неизмененная или незначительно увеличенная толщина стенок желудочков; степень гипертрофии миокарда левого желудочка несопоставима со степенью его дилатации; диффузный характер гипокинезии миокарда; глобальное снижение систолической способности левого желудочка (снижение ФВ и степени систолического укорочения переднезаднего размера левого желудочка, увеличение КСР левого желудочка, снижение сердечного выброса, ударного объема); нарушение сократительной способности миокарда правого желудочка (увеличение его конечного диастолического размера); митральная и трикуспидальная регургитация; наличие внутрипредсердных тромбов (20-28%), у 30-40% больных отмечается косвенный признак тромбоза — спонтанный эхоконтраст в левом предсердии; внутрижелудочковые тромбы чаще встречаются при низкой сократительной способности миокарда. Ранним эхокардиографическим признаком дилатационной кардиомиопатии является увеличение полостей сердца и в первую очередь левого желудочка при отсутствии выраженной гипертрофии миокарда. Остальные эхо-кардиографические признаки появляются позже.
Рентгенологическое исследование
Всегда обнаруживается увеличение размеров сердца преимущественно за счет левого желудочка в начальной стадии заболевания. В последующем наблюдается увеличение всех отделов сердца. В связи с выраженной миогенной дилатацией обоих желудочков сердце приобретает шаровидную форму. Кардиомегалия характеризуется значительным увеличением кардиоторакального индекса (отношение поперечного размера сердца к размеру грудной клетки), который всегда превышает 0,55 и может достигать 0,6 – 0,65.
У большинства больных отмечается нарушение сократительной способности миокарда — сокращения сердца вялые, небольшой амплитуды, часто аритмичные. Обнаруживаются явления венозного застоя в легких, несколько реже — признаки легочной артериальной гипертензии. Застойные явления в легких при рентгенологическом исследовании могут быть выражены умеренно, что объясняется диффузным поражением миокарда обоих желудочков, развитием правожелудочковой недостаточности на ранних стадиях заболевания.
Радионуклидная вентрикулография
Метод основан на регистрации с помощью гамма-камеры импульсов от введенного внутривенно меченного йодом радиоактивного альбумина, проходящего с кровью через левый желудочек. Далее производится компьютерный анализ полученных данных, что позволяет оценить сократительную функцию миокарда, рассчитать объем левого желудочка, фракцию выброса, время циркулярного укорочения волокон миокарда. Радионуклидная вентрикулография обнаруживает увеличение конечного систолического и диастолического объемов ЛЖ, уменьшение ФВ, диффузную гипокинезию ЛЖ. У многих больных выявляется сегментарная асинергия ЛЖ, однако патологических изменений коронарных артерий не обнаруживается.
Сцинтиграфия миокарда
При сцинтиграфии миокарда с радиоактивным таллием 201Т1 могут обнаруживаться мелкие, напоминающие мозаику очаги снижения накопления изотопа, иногда выявляются более крупные дефектные очаги. Дефекты перфузии и накопления изотопа обусловлены множественными очагами фиброза в миокарде, которые развиваются при дилатационной миокардиопатии.
Велоэргометрия
Применяется для дифференциальной диагностики с ИБС и для определения уровня физической работоспособности больного. Характерно значительное снижение толерантности к физической нагрузке, причиной прекращения велоэргометрической пробы являются одышка, усталость, нарушения сердечного ритма, а не боли в области сердца, характерные для больных ИБС.
Рентгеноконтрастная вентрикулография
Позволяет выявить дилатацию желудочков, значительное ослабление их пульсации, диффузную гипокинезию. Участки гипокинезии чередуются с очагами акинезии, что создает диагностические трудности при исключении ИБС. Определяются увеличение КСР и КДР ЛЖ, снижение ФВ. Кличественными ангиокардиографическими критериями дилатационной кардиомиопатии служат индекс конечного диастолического объема ЛЖ свыше 110 см3/м2, индекс конечного систолического объема более 50 см3/м2 и ФВ менее 50%. Характерна умеренная регургитация крови через левое атриовентрикулярное отверстие, иногда визуализируется пристеночный тромб в ЛЖ.
Коронароангиография
При дилатационной кардиомиопатии просвет коронарных артерий не изменен. У некоторых больных наблюдается увеличение количества мелких ветвей коронарных артерий. Предполагается, что это является компенсаторной реакцией, направленной на улучшение кровоснабжения миокарда. Коронароангиография обычно проводится с целью дифференциальной диагностики дилатационной кардиомиопатии и ишемической болезни сердца.
Катетеризация полостей сердца
Этот метод исследования выявляет характерные изменения: значительное увеличение конечного диастолического давления в левом желудочке, а также высокое систолическое и диастоли-ческое давление в легочной артерии и повышение среднего давления в левом предсердии. Конечное диастолическое давление в правом желудочке также увеличено, но степень увеличения значительно меньшая по сравнению с левым желудочком. При развитии правожелудочковой недостаточности отмечается увеличение давления наполнения правого желудочка и среднего давления в правом предсердии.
Морфологическое исследование биоптатов
Прижизненная эндомиокардиальная биопсия является важнейшим инвазивным инструментальным методом диагностики идио-патической дилатационной кардиомиопатии и рекомендуется в комплексном обследовании больных с поражением миокарда неясного генеза с целью дифференциальной диагностики и исключения специфических заболеваний миокарда, имеющих характерные патоморфологические признаки. Чаще всего проводится биопсия миокарда правого желудочка (трансвенозным путем), реже — левого желудочка. В эндомиокардиальных биоптатах отмечаются выраженные дистрофические изменения кардиомиоцитов, явления их некроза, интерстициальный и заместительный склероз различной степени выраженности. Характерно отсутствие активной воспалительной реакции. Нерезко выраженные лимфоцитарные инфильтраты могут встречаться в отдельных участках биоптата, но количество лимфоцитов не превышает 5 или 10 в поле зрения при увеличении микроскопа в 400 и 200 раз соответственно.
КРИТЕРИИ ДИАГНОСТИКИ
Диагностика осуществляется на основании комплексной оценки клинико-инструментальных данных и исключения сходных заболеваний. Диагноз ИДКМП предполагает наличие кардиомегалии с дилатацией полостей сердца, нарушение сократительной функции миокарда; отсутствие известных этиологических факторов, вызвавших развитие дилатационной кардиомиопатии; исключение форм дилатационной кардиомиопатии известной этиологии (ишемической, алкогольной, воспалительной, гипертензивной, клапанной, амилоидной, при системных заболеваниях соединительной ткани, гемохроматозе); исключение экссудативного перикардита. В 1992 г. Р. Manolio разрабртал диагностические критерии ИДКМП.
Большие гемодинамические критерии: ФВ ЛЖ менее 45% или фракционное укорочение переднезаднего размера ЛЖ менее 25% (по данным ЭхоКГ, радионуклидного сканирования, ангиографии) КДР ЛЖ > 117% от предусмотренного значения с учетом возраста и площади тела (> 2,7 см/м2 поверхности тела)
Критерии исключения: артериальная гипертензия (АД > 160 и 100 мм ст. ст.), документированная и подтвержденная повторными измерениями и/или наличием поражения органов-мишеней ИБС (обструкция более 50% диаметра главной коронарной артерии), хроническое употребление алкоголя (ежедневно более 40 г этанола в сутки женщинами и более 80 г мужчинами в течение 5 лет и более, с ремиссией ДКМП после 6 месяцев абстиненции), системные заболевания, перикардиты, легочная гипертензия, врожденные пороки сердца, длительные или пароксизмальные суправентрикулярные аритмии
Диагноз семейной ИДКМП необходимо заподозрить при наличии не менее двух случаев дилатационной кардиомиопатии в одной семье, а также случаев документированной внезапной смерти в возрасте до 35 лет среди родственников первой линии, больных дилатационной кардиомиопатией. Далее диагноз заболевания устанавливается на основании диагностических критериев Н. Mestroni (1999).
Большие диагностические критерии: дилатация сердца, ФВ ЛЖ менее 45% и/или фракционное укорочение переднезаднего размера левого желудочка < 25%
Малые диагностические критерии: необъяснимые суправентрикулярные (фибрилляция предсердий или другие устойчивые аритмии) или желудочковые аритмии в возрасте до 50 лет; увеличение КДР ЛЖ более 117% от рассчитанной нормы с учетом возраста и поверхности тела; необъяснимые нарушения проводимости: атриовентрикулярная блокада 2-3 степени, полная блокада левой ножки пучка Гиса, синоатриальная блокада; необъяснимая внезапная смерть или инсульт в возрасте до 50 лет.
Обсуждая диагностику семейной дилатационной кардиомиопатии, необходимо отметить, что поражение сердца, соответствующее дилатационной кардиомиопатии, может быть первым проявлением нейромышечных заболеваний (мышечные дистрофии Дюшена, Беккера, атаксия Фридрейха). Врач должен помнить о возможности развития дилатационной кардиомиопатии при наследственно обусловленных нейромышечных заболеваниях.
Нейромышечные дистрофии необходимо обязательно исключать в следующих случаях: наличие в семье больных нейромышечными дистрофиями или указание в анамнезе на наличие этих заболеваний у родственников; высокий уровень креатинфосфокиназы в крови; патологические изменения электромиографии; наличие мышечной слабости (особенно прогрессирующей), судорог, мышечной ригидности, псевдогипертрофии голеней.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Дифференцировать идиопатическую дилатационную кардиомиопатию следует с вышеназванными заболеваниями, а также с гипертрофической и рестриктивной кардиомиопатиями, миокардитами. Дифференциальная диагностика идиопатической дилатационной кардиомиопатии будет изложена в последующих разделах при описании других форм кардиопатий.




